Exploring the Inhibitory Potential of Chinese Bittersweet Alkaloid I Against GSK-3β: DFT, Molecular Docking, NCI, and ADME Study
DOI:
https://doi.org/10.62368/pn.v4i1.61Keywords:
GSK-3β, Binding Energy, Pharmacokinetics, molecular docking, NCI Analysis, DFT calculationsAbstract
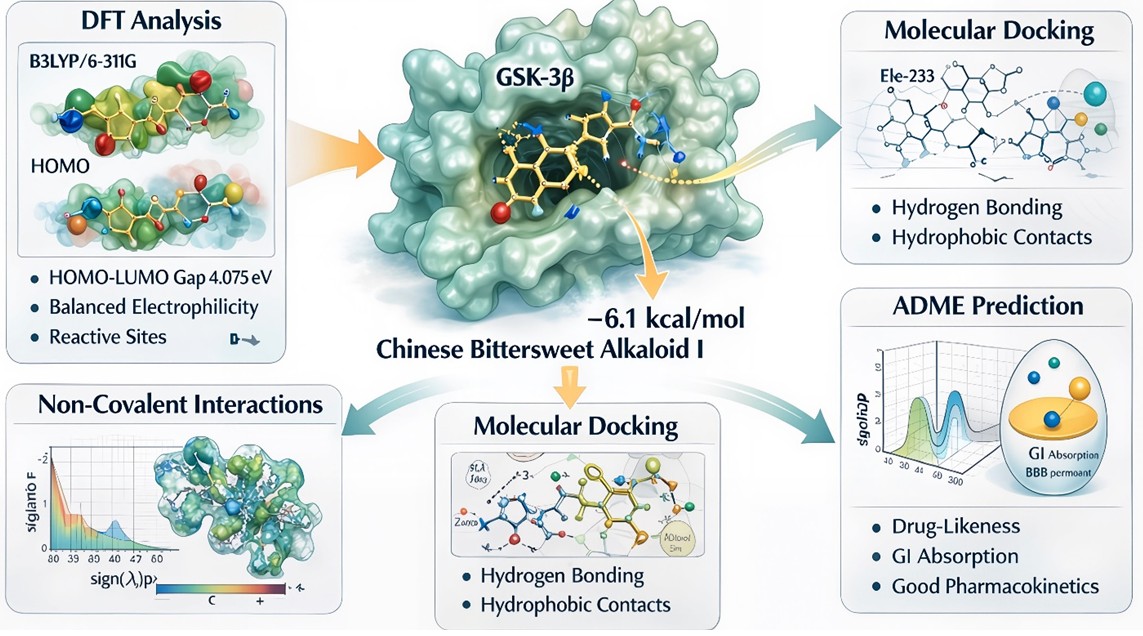
Alkaloids have drawn attention due to their potential role as a therapeutic inhibitor of glycogen synthase kinase-3β (GSK-3β), as it is an essential enzyme associated with neurodegenerative and metabolic disorders. The current study combines Density Functional Theory (DFT), molecular docking, Non-Covalent Interaction (NCI) analysis, and ADME, comprehensively evaluating the inhibitory potential of Chinese Bittersweet Alkaloid I against GSK-3β. Molecular docking revealed a binding energy of –6.1 kcal/mol, showing a favorable interaction stabilized by hydrogen bonding and hydrophobic contacts within the active region of the protein. DFT calculations at the B3LYP/6-311G level provided interpretations into the molecule’s electronic reactivity, with a HOMO–LUMO energy gap of 4.075 eV, balanced electrophilicity, and well-defined reactive sites supported by molecular electrostatic potential mapping. The NCI analysis proved the presence of significant van der Waals and weak attractive interactions taking part into ligand stability. ADME estimation demonstrated good drug-likeness, acceptable pharmacokinetic behavior, and advantageous gastrointestinal absorption, supported by the SwissADME boiled-egg model. These integrated computational results suggest that Chinese Bittersweet Alkaloid I is a potential scaffold for the development of GSK-3β inhibitors, demanding further experimental validation as computational studies inherit limitations.